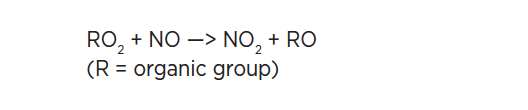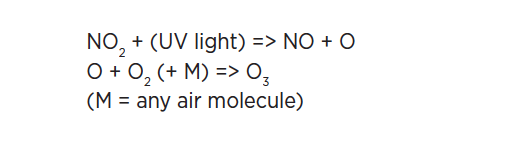As we approach the end of the second decade of the 21st century, changes in atmospheric chemical composition due to anthropogenic pollution continue to challenge the well-being of society. The IUPAC effort in atmospheric chemistry data evaluation can be traced back over 40 years. Global concerns over potentially catastrophic stratospheric ozone depletion resulting from emissions of chlorofluorocarbons (CFCs) led to the creation of the CODATA Task Group on Chemical Kinetics in 1977. The task of the CODATA group was to provide the evaluated kinetic data for atmospheric reactions needed to assess the threat to stratospheric ozone. In 1989, sponsorship of the data evaluation effort was transferred to IUPAC, leading to the formation of the IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation.
Despite greatly improved scientific knowledge of emissions and atmospheric processes over the past 30-40 years, gas-phase and particulate pollution of the atmosphere on local, regional, and global scales continues to be a serious problem. Climate and air quality computer models are important tools which integrate our knowledge of emissions, observations, atmospheric chemistry, and meteorology. Atmospheric models are a critical tool in the formulation of effective policies to address air pollution on all spatial and temporal scales. The IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation provides the evaluated chemical data for atmospheric chemistry used in these models [1].